
The study zeroes in on a biological phenomenon known as cellular senescence, a complex process triggered by various cellular stresses, including DNA damage, telomere shortening, and oncogene activation. In essence, senescent cells undergo a profound transformation: they stop dividing and proliferating, a critical safeguard against cancer, but paradoxically, they do not die. Instead, these "zombie cells," as they are often colloquially known, persist within tissues, becoming active metabolic entities that secrete a potent cocktail of inflammatory molecules, growth factors, and proteases. This collective output is termed the Senescence-Associated Secretory Phenotype, or SASP. The SASP fundamentally alters the local tissue microenvironment, driving chronic low-grade inflammation that can damage surrounding healthy cells and contribute to the pathology of numerous age-related conditions.
"Senescent cells are fairly rare, but think of them like a broken-down car on the 405," explained Anthony Covarrubias, senior author of the study and a distinguished member of the Eli and Edythe Broad Center of Regenerative Medicine and Stem Cell Research at UCLA. "Just one stalled car can back up traffic for miles. Now imagine five or ten of them slowly accumulating. That’s what these cells do to a tissue: even a small number causes enormous disruption." This vivid analogy underscores the disproportionate impact a small population of dysfunctional cells can have on an entire organ system. The continuous release of inflammatory signals from these lingering cells creates a state of chronic sterile inflammation, a hallmark of aging itself, which is increasingly recognized as a foundational driver of conditions ranging from cardiovascular disease and neurodegeneration to metabolic disorders and certain cancers. Understanding and targeting these cellular roadblocks could therefore have far-reaching therapeutic implications.
For years, the scientific community harbored skepticism regarding whether macrophages, the versatile immune cells responsible for patrolling the body, engulfing debris, and presenting antigens, could truly become senescent. Macrophages are renowned for their remarkable plasticity and ability to adapt to diverse tissue environments and functional states. Moreover, healthy, activated macrophages naturally exhibit some molecular features that overlap with those found in senescent cells, such as increased expression of certain inflammatory mediators, making it exceedingly difficult to distinguish between a normally functioning, albeit active, macrophage and one that has entered a detrimental senescent state. This ambiguity hindered progress in understanding their potential role in age-related pathologies.
The UCLA team, however, meticulously addressed this critical challenge by identifying a clear, unambiguous molecular signature for senescent macrophages. Their breakthrough involved pinpointing the concurrent expression of two specific proteins: p21 and TREM2. p21 is a well-established cyclin-dependent kinase inhibitor, frequently upregulated in senescent cells as a key mediator of cell cycle arrest. TREM2, or Triggering Receptor Expressed on Myeloid cells 2, is a cell surface receptor predominantly expressed on myeloid cells, including macrophages and microglia, and plays crucial roles in phagocytosis, lipid metabolism, and inflammation, particularly in neurodegenerative diseases. The researchers discovered that it was the combination of elevated p21 and TREM2 that reliably marked macrophages that were not only truly senescent but also dysfunctional, no longer performing their beneficial immune surveillance and debris-clearing roles, while simultaneously driving chronic inflammation in nearby tissue.
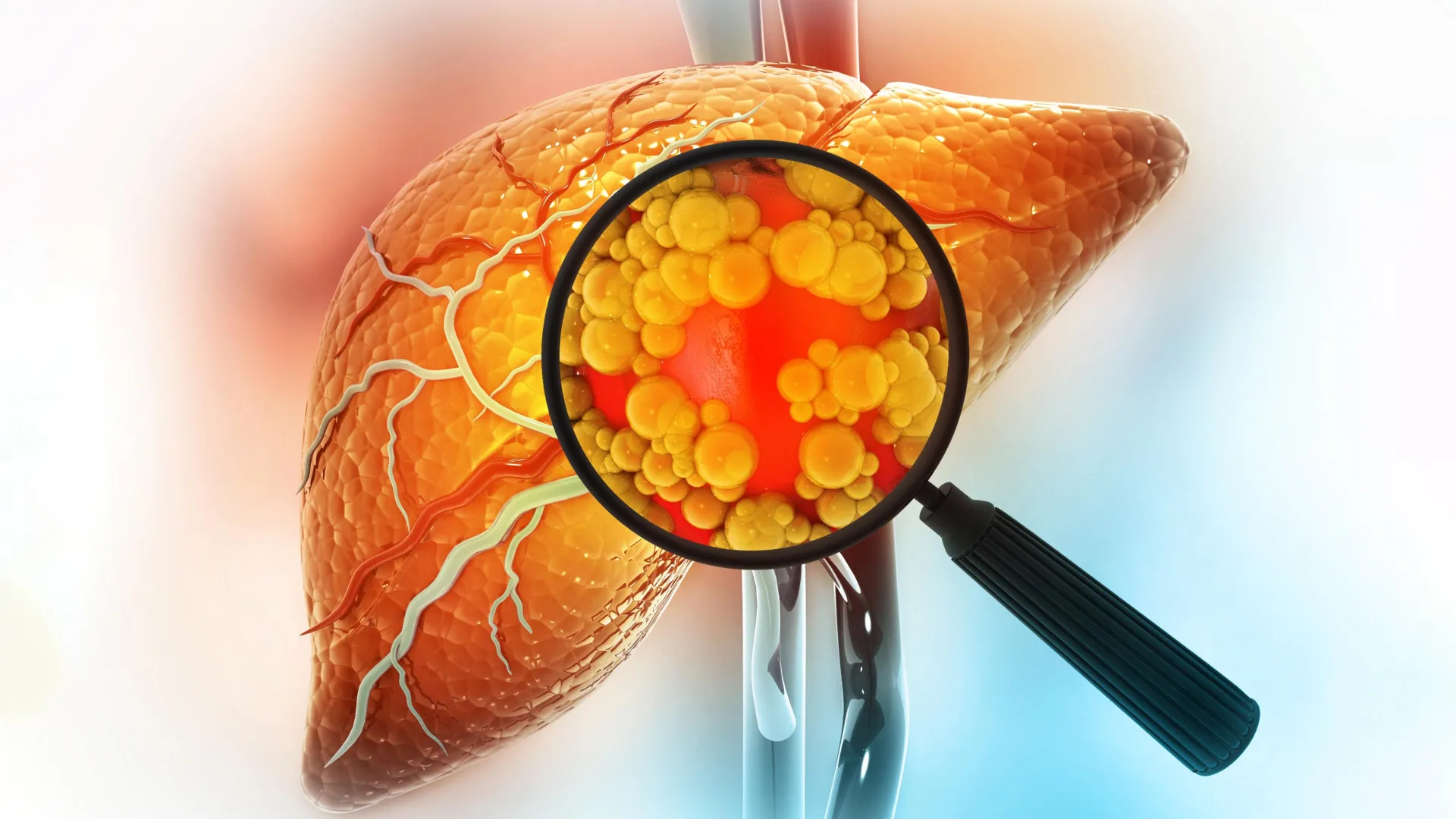
Utilizing this novel and precise molecular marker, the researchers observed a stark and dramatic shift in macrophage populations with advancing age. In the livers of young mice, a healthy approximately 5% of macrophages exhibited the senescent p21-TREM2 signature. In stark contrast, this number skyrocketed in older mice, with a staggering 60% to 80% of liver macrophages displaying the senescent phenotype. This profound age-dependent increase in senescent macrophages correlated precisely with the observed rise in chronic liver inflammation typically seen as animals age, providing compelling evidence for their direct involvement in age-related hepatic decline and systemic "inflammaging."
Aging, however, was not the sole factor contributing to this detrimental cellular buildup. The researchers made another pivotal discovery: excess cholesterol, a ubiquitous component of modern diets and a major risk factor for cardiovascular disease and metabolic syndrome, can also directly push macrophages into a senescent state. To validate this, healthy macrophages were exposed to high levels of low-density lipoprotein (LDL) cholesterol – often termed "bad cholesterol" – in laboratory settings. The results were unequivocal: these macrophages ceased dividing, began actively releasing a torrent of inflammatory proteins characteristic of the SASP, and crucially, displayed the identical p21-TREM2 senescent signature.
"Physiologically, macrophages can handle cholesterol metabolism and are essential for clearing excess lipids," explained Ivan Salladay-Perez, first author of the new study and a graduate student in the Covarrubias lab. "But in a chronic state, it’s pathological. And when you look at fatty liver disease, which is driven by overnutrition and too much cholesterol in the blood, that excess cholesterol appears to be a major driver of the senescent macrophage population." This statement highlights the critical distinction between acute, beneficial macrophage activity and chronic, pathological overload. In the context of persistent high-cholesterol diets, the macrophages become overwhelmed, their lipid-processing machinery dysregulated, leading to cellular stress, eventual senescence, and the perpetuation of inflammation. This insight raises a broader and highly concerning possibility: that diets habitually high in fat and cholesterol may accelerate biological aging by actively promoting macrophage senescence, not just in the liver, but potentially in other vital organs throughout the body, including the brain, heart, and adipose (fat) tissue, thereby contributing to a spectrum of age-related pathologies.
To rigorously test the therapeutic potential of these findings, the UCLA team embarked on a crucial intervention: they treated mice with ABT-263, a pharmacological agent known as a senolytic drug. Senolytics are specifically designed to selectively induce apoptosis (programmed cell death) in senescent cells, thereby clearing them from tissues without harming healthy cells. The effects observed in mice fed a high-fat, high-cholesterol diet were nothing short of dramatic. In untreated animals on the unhealthy diet, livers were significantly enlarged and showed clear signs of severe fatty liver disease, making up approximately 7% of their total body weight. Following treatment with ABT-263, the liver size in these mice dramatically dropped to a healthier 4-5% of body weight. Concurrently, the overall body weight of the treated animals also significantly decreased by about 25%, falling from roughly 40 grams to around 30 grams, indicating a profound metabolic improvement.
Visually, the difference was striking. The livers from the ABT-263-treated mice appeared considerably smaller, healthier, and exhibited a normal reddish color, indicative of reduced fat accumulation and inflammation. In stark contrast, the livers from untreated animals remained visibly enlarged, yellowish, and greasy in appearance – classic hallmarks of severe steatosis (fatty liver). These compelling results unequivocally demonstrate that the removal of senescent macrophages alone can produce major metabolic improvements and reverse significant liver damage, even in the continued absence of dietary changes. "That’s what wowed me," Salladay-Perez remarked. "Eliminating senescent cells doesn’t just slow the fatty liver – it actually reverses it." This finding is particularly impactful, as it suggests a direct therapeutic strategy that could potentially mitigate the devastating effects of unhealthy diets and metabolic dysfunction, even in individuals struggling to adhere to strict lifestyle modifications.
To ascertain the relevance of these mouse-model findings to human health, the researchers meticulously analyzed an existing genomic dataset derived from human liver biopsies. This analysis revealed a highly significant correlation: the same senescent macrophage signature (p21-TREM2) identified in mice was found to be dramatically higher in the diseased livers of human patients compared to healthy human livers. This robust translational evidence strongly suggests that macrophage senescence is not merely an animal model phenomenon but also a significant contributor to the progression and severity of chronic liver disease in humans, including non-alcoholic fatty liver disease (NAFLD) and its more severe inflammatory form, non-alcoholic steatohepatitis (NASH).
The implications of this research are particularly pressing in regions like Los Angeles, where an estimated 30-40% of residents are affected by fatty liver disease, with alarming rates climbing even higher within Latino communities, often linked to genetic predispositions, socioeconomic factors, and dietary patterns. NAFLD and NASH represent a burgeoning public health crisis globally, rapidly becoming the leading cause of liver transplantation. Current treatment options are severely limited, primarily relying on difficult-to-maintain lifestyle changes, and early detection tools for progressive forms of the disease remain woefully inadequate. "This is a huge public health crisis in the making," stated Covarrubias, who is also an assistant professor of microbiology, immunology and molecular genetics. "We’re seeing fatty liver disease in younger and younger people. So we’re really happy to make some inroads into understanding what’s driving it and identifying cell types we might be able to target." The development of targeted therapies that address the underlying cellular mechanisms, such as senescent macrophage accumulation, is therefore of paramount importance.
While ABT-263 proved remarkably effective in mice, its clinical utility in humans is hampered by significant toxicity, including side effects like thrombocytopenia (low platelet count) and neutropenia (low white blood cell count). Consequently, a major focus for the UCLA research team moving forward is to screen for safer, more selective compounds that can effectively eliminate senescent macrophages without eliciting harmful side effects in human patients. This involves high-throughput screening of various small molecules and natural compounds, as well as exploring novel delivery mechanisms to target these cells with greater precision.
Beyond fatty liver disease, the team is actively investigating whether similar pathological processes involving senescent macrophages occur in other age-related diseases. In the brain, for instance, microglia – the resident immune cells and functional equivalents of macrophages in the central nervous system – are known to become dysfunctional with age and in neurodegenerative conditions. It is hypothesized that senescent microglia may contribute to neuroinflammation, the accumulation of amyloid plaques and tau tangles in Alzheimer’s disease, and other forms of neurodegeneration as they encounter and fail to clear large amounts of cellular debris and misfolded proteins. Understanding and targeting senescent microglia could open new therapeutic avenues for these devastating brain disorders. Similarly, senescent macrophages are implicated in atherosclerosis, where they contribute to plaque formation and instability, and in adipose tissue, where they drive chronic inflammation in obesity, further highlighting their widespread impact.
Ultimately, these profound findings provide compelling support for the geroscience hypothesis, a transformative paradigm in biomedical research. This hypothesis posits that by targeting fundamental biological processes of aging, such as cellular senescence, it may be possible to prevent, delay, or treat a broad spectrum of age-related diseases simultaneously, rather than tackling each disease in isolation. In this specific context, the buildup of senescent macrophages, driven by both chronological aging and lifestyle factors like high cholesterol, emerges as a critical, shared mechanism contributing to conditions as diverse as fatty liver disease, atherosclerosis, Alzheimer’s disease, and even certain types of cancer.
"If you really understand the basic mechanisms driving inflammation with aging, you can target those same mechanisms to treat not just fatty liver disease, but atherosclerosis, Alzheimer’s and cancer," affirmed Salladay-Perez. "It all goes back to understanding how these cells arise in the first place." This holistic approach offers a powerful new framework for medical intervention, moving beyond symptom management to address the root causes of age-related pathology. The promise of senolytic therapies, specifically targeting these "zombie" macrophages, could revolutionize how we approach chronic diseases, potentially extending not just lifespan, but more importantly, healthspan – the period of life spent in good health and free from debilitating disease.
This groundbreaking study was made possible through the generous support of key institutions dedicated to advancing biomedical research, including the National Institutes of Health, the Glenn Foundation for Medical Research, the American Federation for Aging Research, and the UCLA-UCSD Diabetes Research Center. Their investment underscores the critical importance of foundational science in uncovering mechanisms that hold the key to future therapeutic breakthroughs.
